Doublet Detection on 10k PBMCs from 10x Genomics v3#
import numpy as np
import doubletdetection
import scanpy as sc
import matplotlib.pyplot as plt
sc.settings.n_jobs=8
sc.set_figure_params()
%matplotlib inline
Download Data from 10x#
Load Count Matrix#
adata = sc.read_10x_h5(
"pbmc_10k_v3_filtered_feature_bc_matrix.h5",
backup_url="https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_filtered_feature_bc_matrix.h5"
)
adata.var_names_make_unique()
# remove "empty" genes
sc.pp.filter_genes(adata, min_cells=1)
Run Doublet Detection#
Here we show-off the new backend implementation that uses scanpy. This new implementation is over 2x faster than version 2.4.0. To use the previous version of DoubletDetection please add the parameters (clustering_algorithm="phenograph", verbose=True, standard_scaling=False) to the classifier and use the thresholds p_thresh=1e-7, voter_thresh=0.8. We recommend using these original parameters if the parameters below lead to undesirable results.
We support the following clustering algorithms:
phenograph
louvain
leiden
The latter two use the scanpy implementations. The default pseudocount for log transform is 0.1. The classifier can become much more memory efficient (but slower) if pseudocount=1 and standard_scaling=False as the array can remain sparse the entire time. This may lead to fewer detected doublets, in which case the prediction thresholds can be manipulated.
clf = doubletdetection.BoostClassifier(
n_iters=10,
clustering_algorithm="louvain",
standard_scaling=True,
pseudocount=0.1,
n_jobs=-1,
)
doublets = clf.fit(adata.X).predict(p_thresh=1e-16, voter_thresh=0.5)
doublet_score = clf.doublet_score()
adata.obs["doublet"] = doublets
adata.obs["doublet_score"] = doublet_score
Visualize Results#
Convergence of doublet calls#
f = doubletdetection.plot.convergence(clf, save='convergence_test.pdf', show=True, p_thresh=1e-16, voter_thresh=0.5)

Doublets on umap#
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata)
sc.tl.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.pl.umap(adata, color=["doublet", "doublet_score"])

sc.pl.violin(adata, "doublet_score")

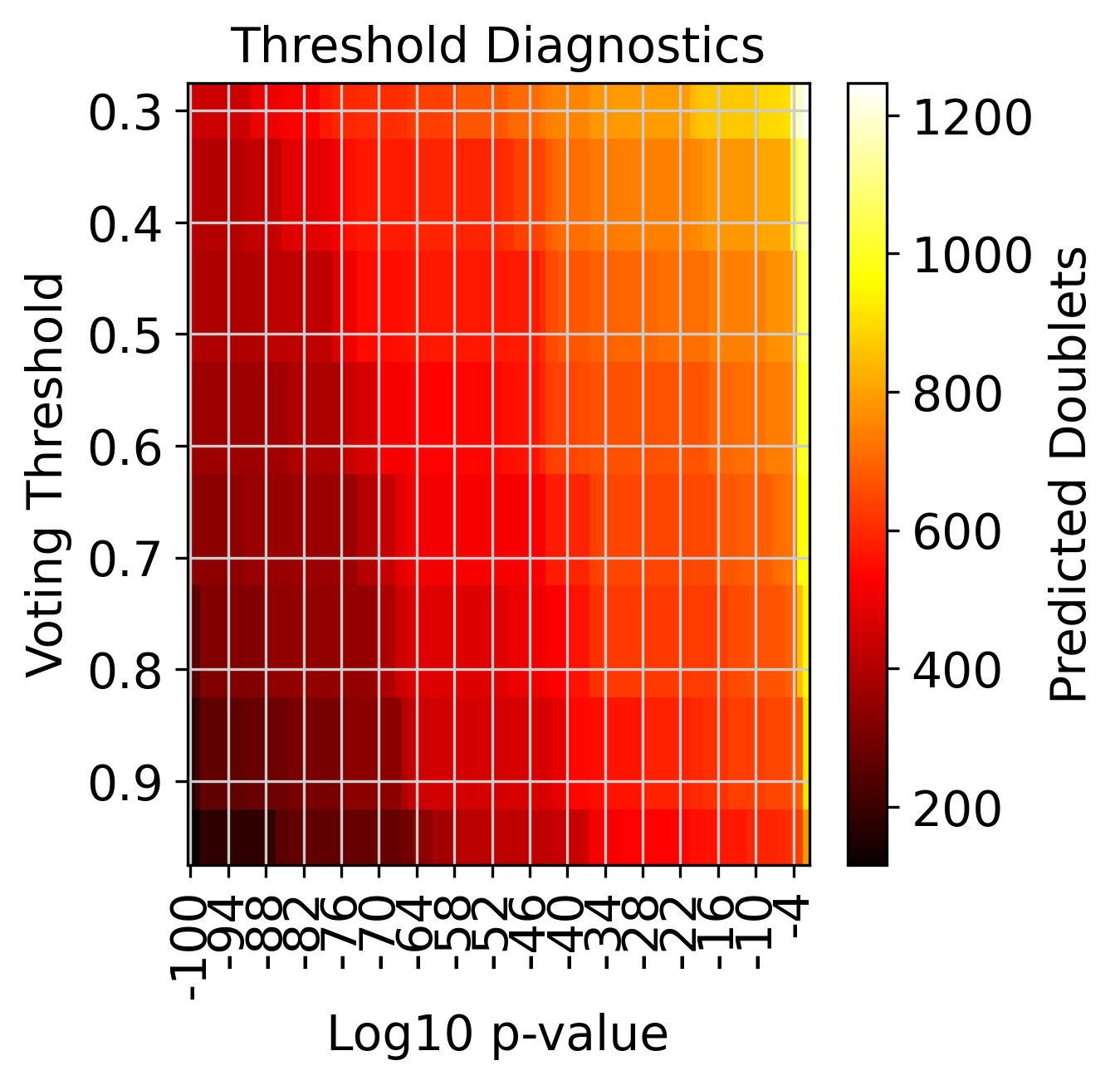
Number of predicted doublets at different threshold combinations#
f3 = doubletdetection.plot.threshold(clf, save='threshold_test.pdf', show=True, p_step=6)